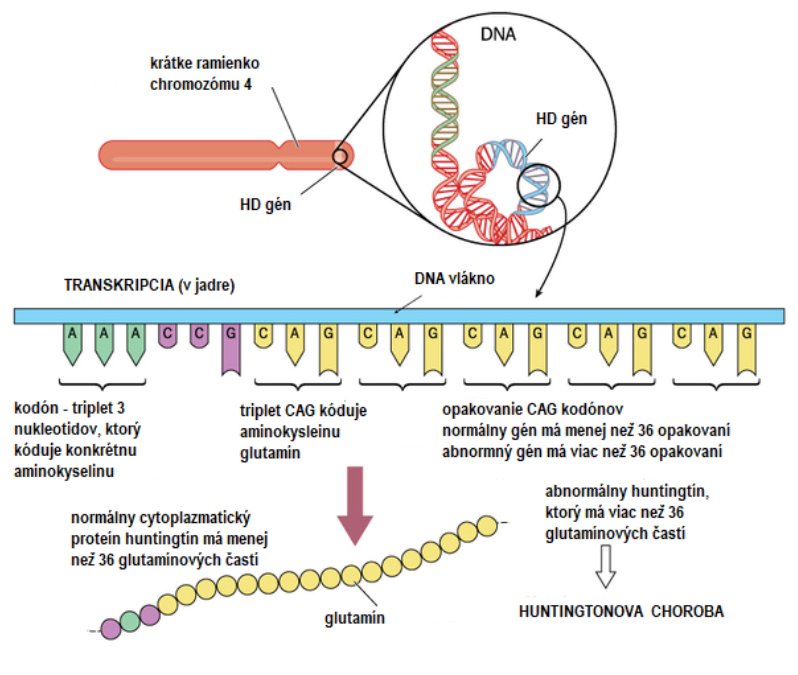
HD je dedičné neurodegeneratívne ochorenie spôsobené mutáciou v huntington géne, ktorá spôsobuje tvorbu proteínu s abnormne dlhými polyglutamínovými sekvenciami.
Fyziologická funkcia proteínu huntingtin nie je úplne známa, avšak zmnožené polyglutamínové sekvencie defektného proteínu sú toxické pre mozgové bunky. Najviac zraniteľné sú neuróny v určitých častiach mozgu a to najmä v bazálnych gangliách (hlavne corpus striatum), ktorých funkcia je dôležitá práve v regulácii motoriky, emócií a kognície. V neskorších fázach ochorenia sú poškodené aj iné regióny mozgu.
V prípade Huntingtonovej choroby mutáciu spôsobuje abnormálne zmnoženie tripletu CAG (triplet ═ trojica nukleotidov, ktoré kódujú aminokyseliny, z ktorých v tele vznikajú bielkoviny), t. j. cytozín-adenín-guanín, ktorý kóduje na chromozóme č. 4 aminokyselinu (glutamín) pre výrobu bielkoviny huntingtín.
Pre ďalšiu prognózu a závažnosť ochorenia je dôležité určiť, o aké až zmnoženie ide.